

在药品生产与质量检测领域,数据完整性与可追溯性是贯穿全流程的核心要求,更是企业符合监管标准、保障药品安全的关键前提。梓梦科技自主研发的显微计数法不溶性微粒仪,凭借精准的检测性能与*的软件管控体系,不仅满足2025版《中国药典》不溶性微粒检测标准,更全面契合《药品记录与数据管理要求(试行)》(国家药-监局2020年发布)及FDA 21CFR第11部分法规要求,为药品检测数据合规提供双重保障。
一、 符合《药品记录与数据管理要求(试行)》的核心体现

《药品记录与数据管理要求(试行)》明确了药品全生命周期中记录与数据的真实性、准确性、完整性和可追溯性要求,尤其对电子记录的系统配置、功能管控、权限管理等作出了细致规定。通过技术设计与软件功能优化,实现多方位合规覆盖。
(一)电子记录完整性与真实性保障
法规要求电子记录需实现原有纸质记录同等功能,确保数据采集、处理、存储全流程可追溯。显微计数法不溶性微粒仪采用全自动扫描检测模式,搭载900万/2000万像素高清数字摄像头,可精准捕捉1-3000μm范围内的不溶性微粒,自动完成颗粒计数、颗粒分布等分析项目,数据采集全程无人工干预,从源头避免手动转录、暂写带来的误差与篡改风险,符合“原始数据应当直接记载于规定记录上"的要求。
显微计数法不溶性微粒仪生成的检测数据可直接按2025版《中国药典》CP0903标准出具报告,包含微粒数量、粒径参数、检测时间、仪器状态等全要素信息,支持数据实时显示、打印与存储,确保电子记录的完整性与可读性,满足法规对电子记录“能够显示所有数据、生成可阅读打印文件"的功能要求。
(二)精细化权限与操作追溯管控
针对电子记录的权限管理,法规强调需建立分级权限体系,确保操作可追溯。显微计数法不溶性微粒仪软件内置用户分级与权限设置功能,可明确区分操作人员、审核人员、系统管理人员等不同岗位权限,业务流程负责人权限与职责严格匹配,有效避免越权操作。同时,系统自动记录所有操作行为,包括用户登录/退出、检测参数设置、数据修改、报告生成等,形成完整的操作日志,清晰标注操作者、操作时间与操作内容,实现操作全程可追溯。
(三)数据安全与备份合规
法规要求电子记录系统需具备稳定的存储与备份机制,确保数据在规定保存期限内可查阅追溯。显微计数法不溶性微粒仪支持数据定期自动备份与手动备份双重模式,备份与恢复流程经过验证,备份记录可全程追溯;系统变更或升级时,可保障原系统数据长期留存与查阅,满足“数据保存期限符合相关规定"的要求。
二、 契合FDA 21CFR第11部分的合规设计
FDA 21CFR第11部分作为药品电子记录与电子签名的核心监管标准,旨在确保电子记录与电子签名的可靠性、真实性与法律效力,与纸质记录和手写签名具备同等地位。显微计数法不溶性微粒仪从系统管控、数据完整性、审计追踪等维度,全面满足其合规要求。

(一)电子记录等效性与不可篡改性
FDA 21CFR第11部分要求电子记录需具备与纸质记录同等的可信度与法律效力,核心在于数据不可篡改与全程可控。显微计数法不溶性微粒仪的检测数据生成后自动加密存储,无法通过非操作修改;软件的审计追踪功能可完整记录数据从生成、修改到删除的行动轨迹,包括隐蔽性操作,确保每一项数据变更都有据可查,杜绝数据伪造与篡改风险,符合法规对电子记录“安全存储、防止篡改"的核心要求。
(二)严格的身份验证与权限管控
法规强调电子记录的操作需建立身份识别与权限分配机制,确保操作责任可追溯。显微计数法不溶性微粒仪通过用户账号与密码实现身份验证,登录信息与操作行为精准关联,形成“一人一账、操作留痕"的管控体系;系统严格区分操作权限与系统管理权限,业务人员无法获取系统管理员权限,避免因权限混淆导致的系统配置变更与数据风险,满足FDA对电子记录操作权限的分级管控要求。
(三)合规性报告与监管适配
FDA 21CFR第11部分要求电子记录需支持监管核查,系统及相关文档需便于FDA检查。显微计数法不溶性微粒仪可生成符合国际标准的中英文检测报告,报告内容涵盖检测方法、仪器参数、数据结果、操作人、审核人等全要素信息,直接适配出口药品检测数据提交需求。
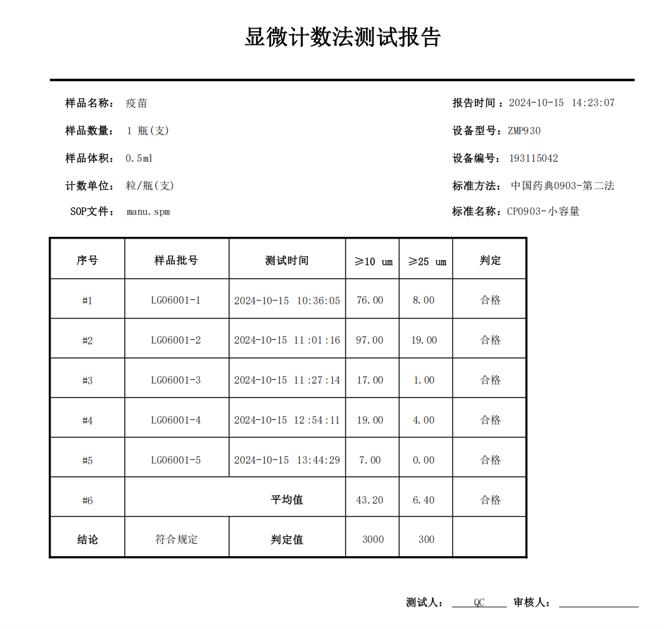
药典报告
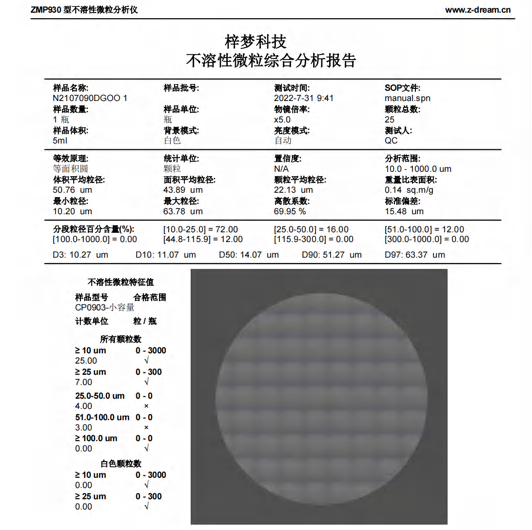
综合报告
药品检测数据的合规性直接关系到药品质量安全与企业市场准入。梓梦科技显微计数法不溶性微粒仪,通过对《药品记录与数据管理要求(试行)》及FDA 21CFR第11部分法规要求的深度适配,构建了从数据采集、处理、存储到追溯的全流程合规体系。同时,依托精准的检测性能与高效的自动化操作,为药品生产企业、检测机构提供“合规+高效+精准"的一体化不溶性微粒检测解决方案,助力企业筑牢药品质量安全防线。
